












Le calcium est l’un des ions les plus souvent a l’origine d’échanges sur les groupes consacrés aux aquariums récifaux. Il est essentiel pour la formation d’une variété de structures, y compris les squelettes et les coquilles de nombreux coraux et autres organismes. Cet article fait parti d’une série avec magnésium et KH à venir. Soyons clairs, cet article bien que long ne pourra pas être complet. Cet article se concentrera sur la nature du calcium dans l’eau de mer et dans nos aquariums, et répondra à des questions telles que «Pourquoi ce dépôt blanc sur mes pompes et chauffages ?” et “Pourquoi mon sable se dissout-t-il?”.
Résumé
Je commence par un résumé ? ehh oui c’est une idée, on verra bien si cela fonctionne. Rien ne vous empêche bien entendu de lire la suite qui est d’un niveau un peu plus élevé. Ici je commence le voyage vers une compréhension loin d’être complète des nombreux aspects du calcium dans les bacs récifaux. C’est un ion compliqué avec une variété d’applications critiques dans les réservoirs récifaux.
Les carbonates sont en sursaturation dans l’eau de nos aquariums. La chaleur des appareils modifie cette sursaturation et un manque de magnésium pour empêcher la précipitation et engendrer un dépôt blanc sur les pompes et chauffages.
Le sable permet un traitement des matières organiques en son cœur, ce traitement engendre une chute de PH ce qui a tendance à le dissoudre.
Bon on a survolé, maintenant si vous vous sentez voila la suite avec les formules. Attention ! Je me suis bien déchiré le cerveau à tenter de comprendre ce que je lisais mais n’étant pas chimiste, il est possible qu’il y ait des erreurs d’écriture, ce qui est important c’est de comprendre le mécanisme.
Calcium dans l’eau de mer
Le calcium est l’un des principaux ions dans l’eau de mer. Il est présent dans l’eau de mer à environ 410 ppm dans le monde entier, il représente donc un peu moins de 1,2% en poids de solides. Les variations de cette concentration sont le plus souvent causées par des changements de salinité, où le calcium monte et descend tout comme la salinité. Une autre cause de variation provient de l’apport des rivières, qui sont souvent fortement enrichies en calcium par rapport à d’autres ions tels que le sodium. Le calcium est également fréquemment enrichi en eau de dégagement hydrothermale qui est libérée sur le fond de l’océan. Le calcium est dissous à partir du basalte chaud lorsque l’eau le traverse et est libéré dans l’océan.
Combien de temps le calcium est il disponible dans la nature
Une façon de se représenter le calcium dans l’eau de mer est de considérer combien de temps un ion de calcium typique est libre en solution avant de précipiter comme un solide tel que le carbonate de calcium. En comparant la concentration connue de l’eau de mer et la quantité ajoutée par les rivières et les évents, on peut estimer le temps de résidence typique du calcium dans l’océan. De même, à partir de la concentration en eau de mer et des taux de sédimentation connus dans l’océan, on peut faire la même chose. Ensemble, ces méthodes produisent une estimation de quelques millions d’années pour le temps de résidence du calcium. En comparaison, un ion à faible renouvellement comme le sodium a un temps de résidence de centaines de millions d’années, et un ion qui est rapidement retiré de la colonne d’eau, tel que l’aluminium, a une résidence d’environ mille ans.
Combien de temps le calcium est il disponible dans l’aquarium
Bien sûr, le temps de séjour du calcium dans les aquariums récifaux est beaucoup plus faible (voir très faible a moins d’une semaine ou moins pour certains bacs), et c’est pourquoi nous y sommes tellement attentifs : si nous ne procédons pas a des ajouts, la concentration diminue rapidement. La même chose ne peut pas être dite pour l’océan. Si tout l’apport de calcium dans l’océan s’arrêtait aujourd’hui, nous ne remarquerions pas la chute du calcium pendant une longue période. La raison de cette différence se résume simplement au très petit volume d’eau dans nos aquariums couplé à un taux élevé de dépôts de carbonate de calcium comparé au volume énorme d’eau dans l’océan couplé à un taux de dépôt moins élevé.
Le calcium en fonction des profondeurs
Un aspect intéressant du calcium dans l’eau de mer est que la concentration en calcium peut être plus élevée dans l’eau profonde que dans l’eau de surface. (Pour nous aquariophiles ça ne sert a rien mais ça permet d’avoir un bon mot lors des réunions de groupes de passionnés) Dans le Pacifique, par exemple, l’eau des mers profondes contient environ 1% de plus de calcium que l’eau de surface. La raison de cette augmentation est que le carbonate de calcium devient beaucoup plus soluble à haute pression, empêchant la précipitation du carbonate de calcium, et permettant même la dissolution des particules de carbonate de calcium formées plus haut dans l’océan qui se déposent dans les profondeurs. La raison de ce changement de solubilité avec pression est assez ésotérique. Lorsque les ions calcium et carbonate sont dissous dans l’eau, un certain nombre de molécules d’eau deviennent étroitement attachés aux ions. Cette hydratation est expliquée en détail dans la section suivante. Dans l’ensemble, cependant, le volume occupé par le carbonate de calcium non dissous et l’eau est plus grand que le volume occupé par les ions calcium et carbonate dissous dans cette même quantité d’eau. Ce changement de volume est principalement dû à la “densité” accrue qui peut être atteinte par les molécules d’eau autour des ions par rapport à l’eau pure. Lorsque cette dissolution se produit sous les énormes pressions exercées sur le fond de l’océan, la dissolution peut en réalité être entraînée par la pression, en raison du changement de volume impliqué, entraînant une plus grande solubilité.
Les précipitations arrivent aussi dans la nature
Enfin, le calcium dans l’océan peut être localement appauvri là où la précipitation du carbonate de calcium est particulièrement rapide. Cela comprend les bancs des Bahamas (où l’aragonite oolithique est précipitée), dans certaines parties de la mer Rouge, et vraisemblablement dans certaines lagunes où la calcification est élevée et le volume d’eau est faible.
État chimique du calcium dans l’eau de mer
Dans de l’eau douce à un pH inférieur à 11, les ions calcium sont essentiellement libres. Autrement dit, ils ne sont fortement attachés à rien d’autre qu’à l’eau et se déplacent indépendamment de tous les autres ions de la solution (sauf si l’eau contient certains agents complexant le calcium, tels que le phosphate ou certains composés organiques). La figure 1 montre un ion calcium hydraté avec des molécules d’eau. Cette sphère d’hydratation est assez fortement attachée à l’ion dans l’eau, avec environ 6-7 molécules d’eau étroitement attachées. Au-delà de cette première sphère d’hydratation, bien sûr, il y a d’autres molécules d’eau dans un arrangement plus lâche, et au-delà, toutes les autres choses en solution. Toutes ces molécules d’eau autour de l’ion s’échangent très rapidement, mais celles qui sont les plus proches de l’ion s’échangent plus lentement et se déplacent avec lui à mesure qu’elles se déplacent dans la solution.
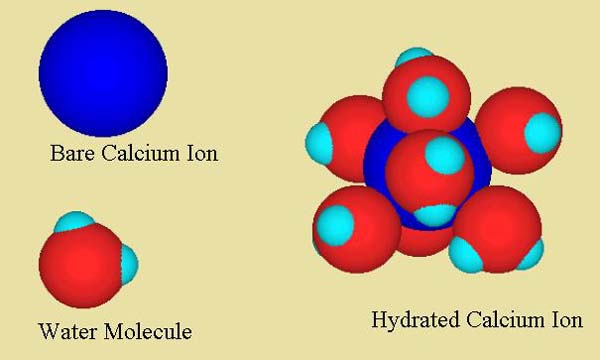
Dans l’eau de mer, la situation est légèrement plus compliquée. Alors que la majorité des ions calcium sont encore libres, certains (environ 10-15%) sont présents sous la forme d’une paire d’ions avec le sulfate, formant la paire d’ions neutres CaSO4 (figure 2). Ces types de paires d’ions solubles sont de courte durée, se formant et se séparant assez rapidement. Néanmoins, ils peuvent avoir un impact significatif sur les propriétés de l’eau de mer. Cette paire d’ions est à son tour hydratée avec des molécules d’eau, comme le montre la figure 2.
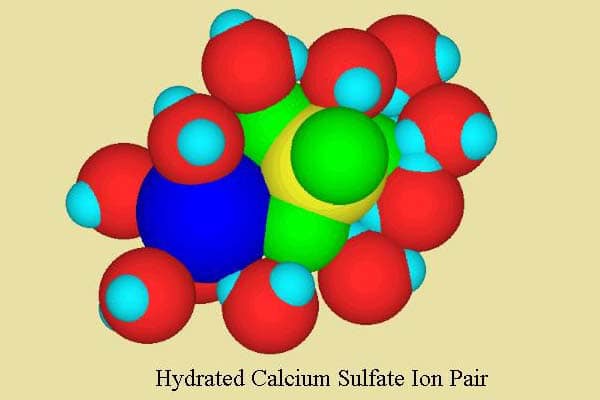
Le calcium forme également des paires d’ions avec le carbonate et le bicarbonate. Alors que ceux-ci comprennent une petite fraction du calcium total, la paire d’ions carbonate de calcium comprend une portion relativement importante du carbonate total (avec du magnésium, environ les 2/3 du carbonate). Ces paires d’ions tendent par conséquent à abaisser la concentration libre de carbonate, et aident ainsi à inhiber la précipitation du carbonate de calcium, et par conséquent à augmenter sa solubilité.
Les appariements du calcium possibles
Enfin, le calcium forme des paires d’ions avec le fluorure, l’hydroxyde, le borate, les diverses formes de phosphate et d’autres ions qui ne sont pas importants pour la concentration en calcium libre, mais peuvent affecter les concentrations libres de ces autres ions. Il se lie par exemple à plus de 70% avec le PO4 —). Dans presque tous les cas, cependant, l’effet du calcium est plus faible que celui du magnésium sur ces ions, parce que la concentration de magnésium est plus élevée et parce que dans certains cas, il interagit plus fortement.
État chimique du calcium dans l’eau de l’aquarium
Le calcium dans l’eau d’un aquarium récifal “devrait” être similaire à celui de l’eau de mer, à quelques exceptions près. En particulier, le calcium est facilement complexé par de nombreux composés organiques et, s’il est présent, peut former des chélates. Certains d’entre eux sont naturels (comme les glucides et les protéines) et peuvent être présents dans des bacs à des niveaux très supérieurs à ceux de l’océan. Des agents liant le calcium supplémentaires sont parfois ajoutés par les aquariophiles, intentionnellement ou non. De tels composés comprennent l’EDTA, l’acide citrique, la vitamine C (acide ascorbique), le polygluconate et les polyphosphates. Par conséquent, une partie des réserves de calcium dans les récifs captifs vont être liée à des composés organiques. Combien et de quel type variera d’un aquarium à l’autre, et peut même ne pas être important dans la beaucoup de cas.
Carbonate de calcium dans l’eau de mer
Un aspect très important du calcium est que, dans l’eau de mer, il est en réalité sursaturé. Ce qui signifie supersaturation dans ce contexte est que, dans les bonnes circonstances, il va précipiter sous forme de carbonate de calcium solide. Bien sûr, dans de nombreuses autres circonstances, il ne le fait pas, et la question de savoir pourquoi est très important. D’abord, quelques définitions. L’expression constante à l’équilibre pour la dissolution du carbonate de calcium est indiquée ci-dessous:
(1) K = [Ca ++] [CO3–]
Lorsque K = Ksp * (le produit de solubilité est constant dans l’eau de mer à n’importe quelle température, pression et salinité), la solution est dite exactement saturée (équation 2)
(2) Ksp * = [Ca ++] [CO3–] (saturation)
Lorsque le produit de la concentration de calcium et de carbonate dépasse le Ksp *, la solution est sursaturée, et il y a “trop” de calcium et de carbonate en solution (équation 3)
(3) Ksp * <[Ca ++] [CO3–] (sursaturation)
Lorsque le produit de la concentration de calcium et de carbonate est inférieur au Ksp *, la solution est censée être sous-saturée, et le carbonate de calcium peut se dissoudre s’il est introduit dans la solution (équation 4)
(4) Ksp *> [Ca ++] [CO3–] (sous-dénaturation)
Dans l’eau de mer normale, l’équation 3 est vraie (sursaturation). Le produit du calcium et du carbonate est environ 3 fois le Ksp* de l’aragonite et 5 fois celui de la calcite (l’aragonite et la calcite sont des formes cristallines différentes du carbonate de calcium). Par conséquent, le carbonate de calcium est prêt à précipiter dans l’eau de mer.
Quels processus empêchent la précipitation du CaCO3 ?
La principale chose qui se passe dans l’eau de mer normale est la présence et donc l’impact du magnésium. Il fait deux choses critiques :
Le magnésium retient les ions carbonate et réduit leur concentration libre, réduisant ainsi la probabilité de précipitation.
Le magnésium pénètre sur la surface du cristal, l’empoisonnant pour éviter la précipitation du carbonate de calcium.
Notez que bien que ces deux processus inhibent la précipitation du carbonate de calcium, le premier augmente réellement la solubilité, tandis que le second ne le fait pas. Il est à noter que la solubilité du carbonate de calcium dans l’eau de mer est environ 26 fois plus élevée que dans l’eau douce à la même température, et ce premier effet du magnésium est l’une des raisons. Le second procédé ci-dessus ne rend pas plus soluble le carbonate de calcium. Dans un sens, il inhibe la voie entre les ions calcium et carbonate solubles, et le carbonate de calcium solide qui se formerait.
D’autres processus qui inhibent la croissance des cristaux dans les aquariums récifaux impliquent à la fois des phosphates et des composés organiques qui pénètrent dans le cristal en croissance, l’inhibant comme le fait le magnésium. Ces processus se produisent vraisemblablement également dans l’eau de mer naturelle, mais comme les concentrations de matières organiques et de phosphate peuvent être plus élevées dans les aquariums, leur effet peut être plus important.
Solubilité du carbonate de calcium dans les aquariums marins
Il y a plusieurs résultats intéressants de la sursaturation du carbonate de calcium dans nos bacs. Un premier que beaucoup de gens rencontrent dans le maintien d’un bac récifal est que les appareils et autres objets chauds (Les chauffages, les pompes, …) semblent être revêtus d’une couche solide blanche au fil du temps. Pourquoi cela ?
Cette couche est principalement du carbonate de calcium, bien qu’il ait probablement d’autres ions (magnésium et autres métaux, phosphate et autres anions, etc.). Les deux raisons pour lesquelles cela arrive sont facilement comprises, mais la principale n’est pas du tout évidente.
Une première raison pour laquelle le carbonate de calcium précipite sur les appareils de chauffage est simplement que le carbonate de calcium est légèrement moins soluble lorsque la température augmente dans l’eau de mer. Puisque le carbonate de calcium est déjà sursaturé, l’effet est que lorsque l’eau est chauffée, la sursaturation du carbonate de calcium augmente, rendant les précipitations plus probables.
La sursaturation (W) du carbonate de calcium dans l’eau de mer est donnée par:
(5) W = [Ca ++] [CO3 -] / Ksp*
Lorsque W = 1, la solution est saturée, et lorsque W> 1, la solution est sursaturée. Plus W est élevé, plus les précipitations sont probables. A une pression de S = 35 et 1 atmosphère, le Ksp* diminue légèrement lorsque la température augmente. Millero (“Chemical Oceanography”, 1996) fournit une série de longues équations pour calculer Ksp* à la fois pour l’aragonite et la calcite. Pour l’aragonite, le log Ksp* chute de -6,19 à 25 ° C, à -6,23 à 40 ° C jusqu’à -6,44 à 80 ° C. En termes relatifs, le KSP* est passé de 1 à 0,91 à 0,55 sur cette plage de température. De même pour la calcite, la valeur de Ksp* relative est passée de 1 à 0,96 à 0,73 sur cette plage.
Par conséquent, si un réservoir a une sursaturation d’environ 3 pour l’aragonite et 5 pour la calcite à 25 ° C (typique pour l’eau de mer dans nos bacs), alors à 40 ° C la sursaturation a augmenté à environ 3,3 et 5,2 , respectivement. A 80 ° C, cette sursaturation a augmenté à 5.4 et 6.8. Avec la sursaturation qui a augmenté, la probabilité de précipitations a augmenté, et cette augmentation fait partie de l’explication de la raison pour laquelle les précipitations ont lieu sur les appareils de chauffage.
Modification de l’acidité de bicarbonate avec la température
Une deuxième raison, peut-être inattendue, à la précipitation du carbonate de calcium sur les objets chauds est liée à la concentration de carbonate. Lorsque l’eau est chauffée, l’équilibre entre le bicarbonate et le carbonate (équation 6) est décalé vers le carbonate.
(6) HCO3- <- -> H + + CO3–
(7) Ka* = [CO3 -] [H +] / [HCO3 -]
(8) [CO3–] = Ka* [HCO3 -] / [H +]
(9) pKa* = -log Ka*
Ce déplacement vers le carbonate est mis en évidence par le déplacement du pKa* de l’eau de mer pour le bicarbonate de 9,00 à 25 ° C à 8,68 à 40 ° C et 8,16 à 80 ° C (calculé à partir des équations fournies par Millero, le * indique simplement qu’il est en l’eau de mer à une température, une pression et une salinité données).
À partir de l’équation 8 (et des dérivations connexes), nous voyons que si le Ka* augmente, alors [CO3–] va augmenter, [H +] va augmenter, et [HCO3-] va diminuer. La question est de savoir combien.
En supposant que la concentration en carbonate soit très inférieure à la concentration en bicarbonate, il à été déterminer la variation de H + avec l’équation 10. L’équation 10 est simplement la solution de l’équation 8 pour H +:
(10) [H +] ~ [Ka* C + Kw*] 1/2
où C est la concentration totale en espèces carbonate / bicarbonate / acide carbonique et pKw* est la constante pour l’auto-dissociation de l’eau. [A propos de l’hypothèse que la concentration de carbonate est inférieure à la concentration de bicarbonate: nous savons que cela est vrai pour l’eau de mer à 25 ° C, mais il est également montré génériquement par Pankow (“Aquatic Chemistry Concepts”, 1991, p. valable avec cette combinaison de pKa * (environ 9), pKw * (environ 13) et C (environ 2 mM) pour d’autres températures également].
En utilisant les valeurs de Ka* et Kw* aux températures appropriées, nous trouvons que [H +] a augmenté d’un facteur d’environ 1,45 entre 25 et 40 ° C. À titre de curiosité, cela a entraîné une diminution du pH d’environ 0,16 unité.
Cependant, ce que nous voulons savoir, c’est la variation de la concentration en carbonate. Pour en revenir à l’équation 8, nous avons:
(11) [CO3 -] 25 = Ka* 25 [HCO3-] 25 / [H +] 25
pour 25 ° C et
(12) [CO3 -] 40 = Ka * 40 [HCO3-] 40 / [H +] 40
pour une eau à 40 ° C. Nous supposons, comme ci-dessus, que [HCO3-] 25 = [HCO3-] 40 (c’est-à-dire que la concentration en bicarbonate est si élevée que la prise d’un peu pour former du carbonate n’affecte pas significativement la concentration en bicarbonate). En substituant le changement connu de Ka* (Ka* 40 = 2.1Ka* 25) et H + ([H +] 40 = 1.45 [H +] 25), on obtient
(13) [CO3 -] 40 = (2,1 ka * 25) [HCO3-] 40 / 1,45 [H +] 25
En combinant les équations 11 et 13, et le fait que [HCO3-] 40 ~ [HCO3-] 25, nous obtenons
(14) [CO3-] 40 = 1,45 [CO3 -] 25
En conséquence, en passant de 25 ° C à 40 ° C, la concentration relative de carbonate a augmenté d’un facteur de 1,45. Nous pouvons maintenant revenir en arrière et confirmer notre hypothèse que le carbonate est encore loin de la concentration en bicarbonate, et c’est clairement le cas, de sorte que cette hypothèse était valide.
Pour en revenir à ce qui nous intéresse vraiment, la sursaturation du carbonate de calcium, nous trouvons que si le carbonate a augmenté d’un facteur de 1,45, alors la sursaturation de la calcite et de l’aragonite a augmenté du même facteur (équation 5).
En effectuant les mêmes calculs pour 80 ° C (pKa = 8,16), nous obtenons une augmentation de la concentration en carbonate d’un facteur de 2,4 par rapport à 25 ° C. Le [H +] augmente également du même facteur.
Comparaison des changements de solubilité et d’acidité sur la sursaturation du carbonate de calcium
En comparant l’augmentation de la sursaturation due aux changements de solubilité et d’acidité entre 25 et 40 ° C pour l’aragonite, nous trouvons un changement de W = 3,0 à 3,3 dû à la solubilité, et de 3,0 à 4,4 dû au changement de bicarbonate pKa. Ensemble, ces effets donnent une sursaturation de 4,8 pour l’aragonite.
De même pour la calcite, le changement de solubilité entre 25 et 40 ° C provoque une augmentation de W de 5,0 à 5,2 pour le changement de solubilité et de 5,0 à 7,3 pour le déplacement du bicarbonate pKa. Ensemble, ces effets donnent une sursaturation de 7,5 pour la calcite.
En comparant l’augmentation de la sursaturation due aux changements de solubilité entre 25 et 80 ° C, nous trouvons un changement de W = 3,0 à 5,4 dû à la solubilité, et de 3,0 à 7,2 dû au déplacement du bicarbonate pKa. Ensemble, ces effets donnent une sursaturation de 13 pour l’aragonite.
De même pour la calcite, le changement de solubilité entre 25 et 80 ° C entraîne une augmentation de W de 5,0 à 6,8 pour le changement de solubilité et de 5,0 à 12 pour le déplacement du bicarbonate pKa. Ensemble, ces effets donnent une sursaturation de 16,3 pour la calcite.
Que signifient vraiment ces valeurs?
Les combinaisons suivantes de calcium et d’alcalinité ont la même sursaturation en eau de mer:
- Eau de mer normale à 80 ° C
- Eau de mer à 25 ° C avec du calcium porté à 1300 ppm
- Eau de mer à 25 ° C avec une alcalinité élevée à 8,2 meq / L
Il est logique que les trois situations ci-dessus puissent conduire à la précipitation du carbonate de calcium, et c’est exactement ce qui se passe sur les surfaces des objets chauds dans nos réservoirs.
Dissolution de CaCO3 dans l’aquarium
Si le carbonate de calcium est sursaturé dans les aquariums marins, comment peut-il également se dissoudre? La réponse réside dans le fait que si la colonne d’eau est sursaturée, d’autres parties du bac peuvent ne pas l’être. Plus précisément, l’eau interstitielle du sable et de la roche est souvent inférieure au pH de la colonne d’eau. Par exemple, si je mets une sonde de pH dans un lit de sable d’aragonite oolitique, j’obtiens un pH dans les 7, quand la colonne d’eau elle-même a un pH = 8,4. (j’avoue je n’y ai pas crus et j’ai testé et c’est vrai)
Les raisons pour lesquelles le pH est plus bas dans le sable sortent du cadre de cet article, mais elles concernent la décomposition des composés organiques (et de certains composés azotés). L’oxydation aérobie et anaérobie des composés organiques dans l’eau de mer peut conduire à la production d’acide, en particulier d’acide carbonique dérivé du CO2. Ce lien montre certaines des réactions qui peuvent (et ne peuvent pas) produire de l’acide dans les lits de sable.
Lorsque le pH est abaissé, l’équilibre entre le carbonate et le bicarbonate se déplace vers le bicarbonate (c’est-à-dire vers la gauche dans l’équation 15):
(15) HCO3- <- -> H + + CO3–
Ce changement réduit considérablement la concentration en carbonate. En utilisant l’équation 8, on peut calculer que la concentration de carbonate diminue d’environ un facteur de 3 pour une chute de pH de 0,5 et d’un facteur de 10 pour une chute de pH totale. En conséquence, l’aragonite devient d’abord soluble dans l’eau de mer lorsque le pH descend en dessous d’environ 7,7 (cette valeur pourrait être plus proche de 7,5-7,7 dans les bacs récifaux où l’alcalinité est souvent plus élevée que dans l’eau de mer). Ce niveau est atteint dans certains lits de sable et permet la dissolution d’une partie du sable.
La vitesse de dissolution est cependant assez faible, car le taux de libération et de dégradation des composés organiques (ou de certains composés azotés) suffisamment profond dans le sable pour permettre une chute du pH est relativement faible. Le débit variera cependant d’un réservoir à l’autre, car les différentes façons de distribuer les matières organiques dans les parties plus profondes du sable varieront (diffusion, mouvement par les organismes, mort des organismes, etc.). Notez que la nécessité d’oxyder les matières organiques dans les parties plus profondes du sable pour permettre la dissolution du sable n’a rien à voir avec l’oxygénation du sable. Cela a plus à voir avec le fait qu’à proximité des surfaces du sable, le pH sera plus proche de celui de l’eau du bac par transfert d’acide et de base de la colonne d’eau, et il faut être assez profond pour permettre un pH plus bas de s’établir.
References
Crystal growth of calcium carbonate in artificial seawater: The effect of temperature and of the presence of inhibitors. Kladi, Elina; Ostvold, Terje; Klepetsanis, Pavlos G.; Amjad, Zahid; Koutsoukos, Petros G. FORTH-ICEHT, Patras, Greece. Book of Abstracts, 218th ACS National Meeting, New Orleans, Aug. 22-26 (1999).
Solubility of calcite in seawater solution of different magnesium concentrations at 25°C and 1 atm total pressure: a laboratory re-examination. Rushdi, Ahmed I.; Chen, Chen-Tung Arthur; Suess, Erwin. Coll. Oceanic Atomospheric Sci., Oregon State Univ., Corvallis, OR, USA. Umi (1998), 36(1), 9-22.
The problem of alkaline scale formation from a study on Arabian Gulf [Persian Gulf] water. El Din, A. M. Shams; Mohammed, Rizk A. Mater. Test. Lab., Water Electr. Dep., Abu Dhabi, United Arab Emirates. Desalination (1989), 71(3), 313-24.
Apparent calcite supersaturation at the ocean surface: why the present solubility product of pure calcite in seawater does not predict the correct solubility of the salt in nature. Cooke, Robert C.; Kepkay, Paul E. Dep. Oceanogr., Dalhousie Univ., Halifax, NS, Can. Mar. Chem. (1984), 15(1), 59-69.
Calculation of saline waters supersaturation on calcium carbonate, magnesium hydroxide and calcium sulfate, taking into account ion associates formation. Martynova, O. I.; Vasina, L. G.; Krotova, I. S. Inst. Power Eng., Moscow, USSR. Proc. Int. Symp. Fresh Water Sea (1976), 1 329-36.
Calcium carbonate retention in supersaturated sea water. Pytkowicz, R. M. Sch. Oceanogr., Oregon State Univ., Corvallis, Oreg., USA. Amer. J. Sci. (1973), 273(6), 515-22.
Inhibition of aragonite precipitation from supersaturated sea water: a laboratory and field study. Berner, Robert A.; Westrich, Joseph T.; Graber, Ron; Smith, Jennifer; Martens, Christopher S. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Am. J. Sci. (1978), 278(6), 816-37.
Biologically mediated dissolution of calcium carbonate above the chemical lysocline? Milliman, J. D.; Troy, P. J.; Balch, W. M.; Adams, A. K.; Li, Y.-H.; Mackenzie, F. T. School of Marine Science of the Virginia Institute of Marine Science, College of William and Mary, Gloucester Pt., VA, USA. Deep-Sea Res., Part I (1999), 46(10), 1653-1669.
Excess dissolved Ca in the deep ocean: a hydrothermal hypothesis. De Villiers, Stephanie. Department of Geological Sciences, University of Washington, Seattle, WA, USA. Earth Planet. Sci. Lett. (1998), 164(3-4), 627-641.
Evidence in support of first-order dissolution kinetics of calcite in seawater. Hales, Burke; Emerson, Steve. Lamont-Doherty Earth Observatory of Columbia University, Palisades, NY, USA. Earth Planet. Sci. Lett. (1997), 148(1-2), 317-327.
The crystallization of calcium carbonate in artificial seawater; role of the substrate. Sabbides, Theophylaktos G.; Koutsoukos, Petros G. Institute of Chemical Engineering and High Temperature Chemical Processes, and Department of Chemical Engineering, University of Patras, P.O. Box 1414, Patras, Greece. J. Cryst. Growth (1993), 133(1-2), 13-22.
Possible role of carbonate dissolution in estuarine phosphate dynamics. De Jonge, V. N.; Villerius, L. A. Tidal Waters Div., Rijkswaterstaat, Haren, Neth. Limnol. Oceanogr. (1989), 34(2), 332-40.
Synthetic magnesian calcites solubilities in seawater as a function of pressure. Koch, B. Lab. Oceanol., Univ. Liege, Sart Tilman, Belg. Oceanol. Acta (1985), 8(4), 413-21.
Kinetics of dissolution and precipitation of calcium carbonate in seawater. Berner, Robert A. Dep. Geol. Geophys., Yale Univ., New Haven, CT, USA. Crist., Deform., Dissolution Carbonates, Reun. (1980), 33-57.
The solubility of calcium carbonate in sea water at 2?C based upon in-situ sampled pore water composition. Sayles, Frederick L. Woods Hole Oceanogr. Inst., Woods Hole, MA, USA. Mar. Chem. (1980), 9(4), 223-35.
The thermodynamics of the carbonate system in seawater. Millero, Frank J. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, FL, USA. Geochim. Cosmochim. Acta (1979), 43(10), 1651-61.
The effect of pressure on the solubility of calcite in seawater at 25?C. Millero, Frank J. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, Fla., USA. Geochim. Cosmochim. Acta (1976), 40(8), 983-5.
Dissolution kinetics of calcium carbonate in seawater: VII. The dissolution kinetics of synthetic aragonite and pteropod tests. Morse, John W.; De Kanel, John; Harris, Karen. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, FL, USA. Am. J. Sci. (1979), 279(5), 488-502.
Dissolution kinetics of calcium carbonate in sea water. VI. The near-equilibrium dissolution kinetics of calcium carbonate-rich deep sea sediments. Morse, John W. Rosenstiel Sch. Mar. Atmos. Sci., Univ. Miami, Miami, Fla., USA. Am. J. Sci. (1978), 278(3), 344-53.
Dissolution kinetics of calcium carbonate in sea water. V. Effects of natural inhibitors and the position of the chemical lysocline. Morse, John W. Dep. Oceanogr., Florida State Univ., Tallahassee, Fla., USA. Amer. J. Sci. (1974), 274(6), 638-47.
Dissolution kinetics of calcium carbonate in sea water. IV. Theory of calcite dissolution. Berner, Robert A.; Morse, John W. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Amer. J. Sci. (1974), 274(2), 108-34.
Dissolution kinetics of calcium carbonate in sea water. III. New method for the study of carbonate reaction kinetics. Morse, John W. Dep. Oceanogr., Florida State Univ., Tallahassee, Fla., USA. Amer. J. Sci. (1974), 274(2), 97-107.
Dissolution kinetics of calcium carbonate in sea water. II. Kinetic origin for the lysocline. Morse, John W.; Berner, Robert A. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Amer. J. Sci. (1972), 272(9), 840-51.
Dissolution kinetics of calcium carbonate in sea water. I. Saturation state parameters for kinetic calculations. Berner, Robert A.; Wilde, Pat. Dep. Geol. Geophys., Yale Univ., New Haven, Conn., USA. Amer. J. Sci. (1972), 272(9), 826-39.
Chemistry and the Aquarium: Calcium By Randy Holmes-Farley http://www.advancedaquarist.com/2002/3/chemistry





 pas la fin")




{kind=link}